
Rabema Manual
Introduction
Rabema can be used in two modes: You can both build a gold standard for real-world reads (“normal mode”) or use simulated data where the original sample position (“oracle mode”) is known. Of course, the real-world mode can also be used for simulated reads when ignoring the information about the original sample position. In order to use follow this tutorial, you will also need to install samtools for sorting BAM files. The tutorial assumes you are using Linux or Mac Os X.
Using Rabema In Normal Mode
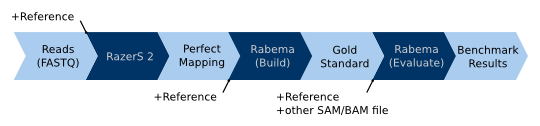
The following picture shows the typical normal Rabema workflow for real-world and simulated data without sample position
information: (1) The reads and the reference sequence are passed to a fully sensitive read mapper, e.g. RazerS
(version >= 2). The result is a “perfect” mapping with one match per interval/lake. (2) This perfect mapping is then
passed to Rabema (rabema_build_gold_standard) to build a gold standard intervals (GSI) file. (3) This GSI file can now
be used to evaluate the output of an arbitrary read mapper. Together with the reference sequence and the read mapper
result as a SAM or BAM file, the GSI file is passed to Rabema (rabema_build_gold_evaluate) to perform the evaluation.
Steps (1) and (2) only have to be performed once for every data set (reference and reads file). Step (3) is repeated for
each output of a read mapper to evaluate.
(1a) Building the Perfect Mapping
First, we have to build the perfect mapping from a reference FASTA file and a reads file (FASTA or FASTQ). The output is a SAM file that contains (at least) one match for each Rabema lake. We use RazerS 3 for this:
razers3 -v -rr 100 -i 92 -m 1000000 -ds \
-o gold_pre.sam genome.fa reads.fq
We are using the following command line arguments:
-vEnable verbosity.-rr 100Set recognition rate to 100%, lossless mode.-i 92Set identity to 92%, i.e. error rate is 8%.-m 1000000Collect up to 1M alignments per read. This is just an arbitrary, large enough number. No read should have remotely that many SAM records.-dsDo not shrink alignments, required for full sensitivity in terms of Rabema benchmark.-oOutput file is “gold.sam”- The last two arguments are the path to the reference FASTA file and the reads FASTQ file.
(1b) Postprocessing the Perfect Mapping
We have to postprocess this SAM file. The output of RazerS only includes the read sequence and qualities for once record
for each read (as described in the “best practices” of the SAM 1.4 specification). Rabema comes with a program
rabema_prepare_sam that accepts a SAM file that is sorted by read name and fills the read sequence and qualities into
each SAM file.
rabema_prepare_sam -i gold_pre.sam -o gold_by_qname.sam
To build the gold standard, we have to sort the updated file by coordinate. The simplest way is to convert it to BAM and
then use samtools for sorting.
samtools view -Sb gold_by_qname.sam >gold_by_qname.bam
samtools sort gold_by_qname.bam gold_by_coordinate
The resulting BAM file gold_by_coordinate.bam can now be used by Rabema to build the gold standard.
(2) Building the Gold Standard
The gold standard is built by the program rabema_build_gold_standard:
rabema_build_gold_standard -e 8 -o gold_standard.gsi \
-r genome.fa -b gold_by_coordinate.bam
We have to set the following parameters:
-e 8We use an error rate of 8%. This has to be the same error rate as we built the gold standard with (although, there the inverse (identity) is used).-o gold_standard.gsiThe output gold standard intervals (GSI) file.-r genome.faThe reference FASTA file.-b gold_by_coordinate.bamThe perfect mapping, sorted by coordinate we generated earlier. Note that we could also pass a SAM file using the-s/--in-samparameter. Optionally, we can use--distance-metric hammingto use Hamming distance instead of edit distance.
(3) Read Mapper Evaluation
We can now use the file gold_standard.gsi to evaluate the result of any read mapper producing the SAM output of read
mappers with the program rabema_evaluate. Say, the read mapper’s output is result.sam. This file must be sorted by
read name.
We now call rabema_evaluate as follows. Logging is written to stderr, a result report is written to stdout.
rabema_evaluate -c all-best -e 8 -r genome.fa \
-g gold_standard.gsi -b result.bam
We are using the following parameters:
-c all-bestThe benchmark category to use (defaults to “all”)-e 8The error rate to evaluate the read mapper with (defaults to 0).-r genome.faThe reference sequence.-g gold_standard.gsiThe gold standard file to use.-b result.bamThe read mapper’s resulting BAM file. (Again, we could use SAM using the-s/--in-samparameter). Note that the error rate to use for the evaluation does not have to be equal tot he error rate when generating the gold standard. Valid values for the benchmark category are “all”, “all-best”, and “any-best”.
Using Rabema In Oracle Mode
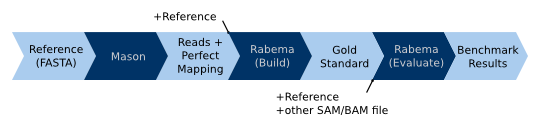
When using simulated data with origin information in oracle mode, the read simulator replaces the fully sensitive read
mapper in the workflow for real-world data. (1) Read simulators such as Mason already generate a SAM
file with the original sample location together with the reads FASTA or FASTQ file. We only have to sort the file with
the original sample location by coordinate. (2) This sorted SAM/BAM file is then passed to Rabema to build the gold
standard. We use Rabema in oracle mode, meaning that the SAM file contains exactly one record for each read with the
correct sample location. Rabema will only construct correct intervals from the gold standard. Each interval will be
expanded with the error rate of the read alignment (to the reference) at the original sample location. This generates a
gold standard intervals (GSI) file. (3) This file is then passed together with the reference sequence and the output of
an arbitrary read mapper to Rabema (rabema_build_gold_evaluate) to perform the benchmark.
(1a) Simulating Read Using Mason
We use the read simulator Mason for generating the reads FASTQ sim.fq and the SAM file with the sample locations
sim.sam.
mason illumina -sq genome.fa
mv genome.fa.fastq sim.fq
mv genome.fa.fastq.sam sim.sam
We use Mason to simulate read using a simple Illumina-like error mode. The parameter -sq tells Mason to simulate qualities. The resulting SAM file is now sorted by read id but we need it sorted by coordinate.
(1b) Sorting Perfect Mapping SAM File
Again, the simplest way for sorting is using samtools.
samtools view -Sb sim.sam >sim.bam
samtools sort sim.bam sim_by_coordinate
(2) Building Gold Standard In Oracle Mode
We now pass it the file sim_by_coordinate.bam to Rabema to build the GSI file in oracle mode. Note that we do not need to pass an error rate in oracle mode because for each interval, the error rate at the sample position is used.
rabema_build_gold_standard --oracle-mode -o sim.gsi \
-r genome.fa -b sim_by_coordinate.bam
(3) Read Mappper Evaluation In Oracle Mode
We can now run the evaluation step on a read mapper output file result.sam using Rabema in oracle mode. In this mode,
the specified benchmark category and error rate is ignored because there is exactly one interval for each read that has
to be found. Again, the result file must be sorted by read name.
rabema_evaluate -r genome.fa -g sim.gsi -b result.bam
Optionally, we could also use the SAM format for the read mapper’s result using the -s/--in-sam parameter.
Using Rabema With Paired-End Data in Oracle-Mode
Rabema can also be sensibly used with paired-end data in oracle mode. First, we simulate paired-end reads using Mason.
mason illumina -sq -mp genome.fa
mv genome.fa_1.fastq sim_1.fq
mv genome.fa_2.fastq sim_2.fq
mv genome.fa.fastq.sam sim.sam
Second, we sort the reads by coordinate:
samtools view -Sb sim.sam >sim.bam
samtools sort sim.bam sim_by_coordinate
Next, we build the gold standard using Rabema in oracle mode. Rabema will automatically recognize that the data is paired-ended from the SAM file.
rabema_build_gold_standard --oracle-mode -o sim.gsi \
-r genome.fa -b sim_by_coordinate.bam
Finally, we can compare read mapper results against the build gold standard.
rabema_evaluate -r genome.fa -g sim.gsi -b result.bam
Frequently Asked Questions
Q: Which SAM fields are required when evaluating a read mapper?
The fields QNAME, FLAG, RNAME, POS, CIGAR, and SEQ are interpreted by Rabema. Usually, all of them except SEQ are present in read mapper output by default. You can add the sequence using the program rabema_prepare_sam as described above. Note that the SAM file must be valid. Some read mappers produce invalid SAM files (i.e. invalid flags or tags).
Q: What if a read mapper stores secondary alignments in the XA tag?
Read mappers such as BWA and Hobbes only write one SAM record. The Rabema distribution contains a Python script
expand_xa.py that allows to expand the XA tag into one record for each entry.
Use it as follows: expand_xa.py <IN.sam >OUT.sam.
Q: What if the CIGAR field is missing?
Read mappers such as Hobbes only write the CIGAR field if configured so on the command line. Use the appropriate switch to enable the generation of the CIGAR field.
Q: Can I use RABEMA for paired-end data?
Paired-end reads can only be used in oracle mode, i.e. with known origin location. However, since the core of the read alignment problem is that of finding all locations for each mate, benchmarking single-end alignments is still very useful. Some words on the difficulty of creating a gold standard formulation for paired-end read alignment: There is no obvious extension of the Rabema single-end mode to paired-end mode and detailed investigation remains future work. When aligning paired-end data, one opens many more degrees of freedom than for single-end mode. First, a question is whether to consider concordantly aligning reads (i.e. with correct orientation and having an insert size within expected bounds) only or whether discordant reads are allowed. If only concordant mapping locations are to be found then we end up with a two-criteria optimization problem. Given multiple feasible alignment location for each end of the pair that could be combined to form a concordantly mapping, which one is to pick? The one closest to the the expected or mean insert size? The one with the best individual mappings?